Glycan-deficient PrP stimulates VEGFR2 signaling via glycosaminoglycan
Date:02-06-2016 | 【Print】 【close】
One of the most common protein post-translational modifications is N-linked glycosylation. N-linked glycosylation is important in proper protein folding, transit to the cell surface and secretion, in a protein, as well as cell context dependent manner. The normal cellular prion protein, PrP, is a widely expressed, highly conserved, glycosylphosphatidylinositol (GPI)-anchored cell surface glycoprotein. The two N-linked glycosylation sites on human PrP are located at the C-terminus at residues 181 and 197. A plethora of more than 40 proteins have been reported to bind PrP. In addition, PrP also binds divalent cations, such as copper and zinc, lipids, nucleic acids and GAG. However, other than being essential for the pathogenesis of prion diseases, the normal functions of PrP remain obscure; Prnp−/− mouse is normal without apparent phenotype. Whether the two glycans are important in PrP biology is debated. In some cell models, glycan-deficient PrP is retained in the cytosol, unable to reach the cell surface. On the contrary, other studies find them to be dispensable for cell surface expression. Even though the two N-linked glycans are not essential for the pathogenesis of prion diseases, they do contribute to disease phenotypes as well as strain specificity of some infectious prion. However, the underlying mechanisms by which N-linked glycans modulate disease phenotype or strain specificity are not known.
Because of these ambiguities, the Research Group of Prion Cell Biology led by Prof. Chaoyang Li investigated whether the two N-linked glycans were important in PrP biology using CHO cell, which lacked detectable endogenous hamster PrP. They transfected a wild type (WT) human PrP; a single glycan site mutant (SM) PrP, in which one of the glycosylation sites, asparagine (N) 181, was altered to an alanine (A) (N181A); another SM PrP, in which the other site, N197, was mutated to an A (N197A); a double mutant (DM) PrP, in which both sites were replaced with A individually into CHO cells. These mutated PrP are collectively referred to as glycan-deficient PrP. They find that all glycan-deficient PrP are expressed on the cell surface in levels comparable to WT PrP. However, glycan-deficient PrP appears to partition more in lipid raft than wild-type PrP. Furthermore, glycan-deficient PrP forms complex with VEGFR2 and GAG resulting in the activation of VEGFR2 and the PI3K-Akt signaling pathway. These biochemical alterations also resulted in changes in cellular behavior; compared to cells with wild type PrP, cells with glycan-deficient PrP are more adhesive and more mobile. Collectively, these results suggest that in CHO cells, one of the functions of N-linked glycans on PrP is to negatively regulate PrP functions.
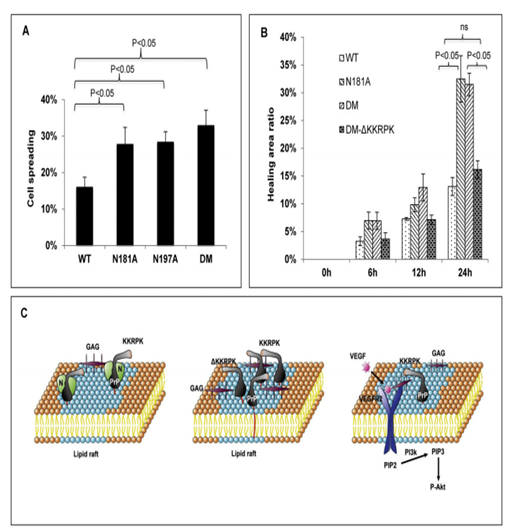
MG and UG PrP cells tend to spread and migrate faster than WT PrP cells.